
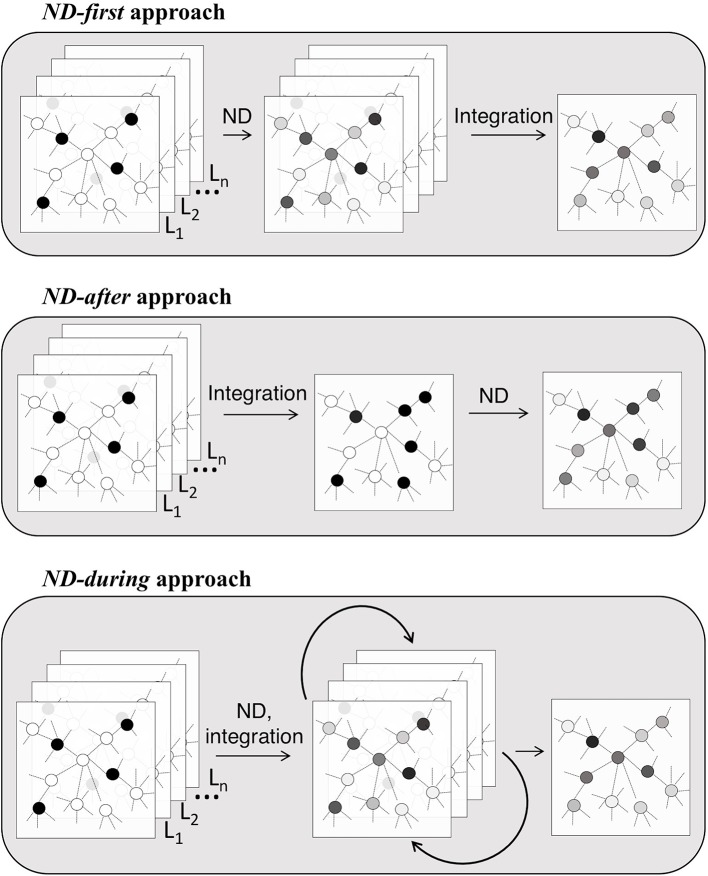
The event of integrative strategies is among the predominant challenges in bioinformatics. Community-based strategies for the evaluation of a number of gene-centered datasets have in mind recognized and/or inferred relations between genes.
Within the final many years, the mathematical equipment of community diffusion-also known as community propagation-has been exploited in a number of network-based pipelines, because of its capacity of amplifying affiliation between genes that lie in community proximity.
Certainly, community diffusion gives a quantitative estimation of community proximity between genes related to a number of completely different information sorts, from easy binary vectors to actual vectors. Subsequently, this highly effective information transformation methodology has additionally been more and more utilized in integrative analyses of a number of collections of organic scores and/or a number of interplay networks.
We current an outline of the state-of-the-art of bioinformatics pipelines that use community diffusion processes for the integrative evaluation of omics information. We focus on the basic methods wherein community diffusion is exploited, open points and potential developments within the discipline.
Present tendencies recommend that community diffusion is a device of broad utility in omics information evaluation. It’s cheap to suppose that it’s going to proceed for use and additional refined as new information sorts come up (e.g. single cell datasets) and the identification of system-level patterns can be thought-about increasingly more essential in omics information evaluation.
Community Diffusion Promotes the Integrative Evaluation of A number of Omics.
Multi-omic evaluation of gametogenesis reveals a novel signature on the promoters and distal enhancers of energetic genes.
Epigenetic regulation of gene expression is tightly managed by the dynamic modification of histones by chemical teams, the variety of which has largely expanded over the previous decade with the invention of lysine acylations, catalyzed from acyl-coenzymes A. We investigated the dynamics of lysine acetylation and crotonylation on histones H3 and H4 throughout mouse spermatogenesis. Lysine crotonylation seemed to be of great abundance in comparison with acetylation, notably on Lys27 of histone H3 (H3K27cr) that accumulates in sperm in a cleaved type of H3.
We recognized the genomic localization of H3K27cr and studied its results on transcription in comparison with the classical energetic mark H3K27ac at promoters and distal enhancers.
The presence of each marks was strongly related to highest gene expression. Evaluation of their co-localization with transcription regulators (SLY, SOX30) and chromatin-binding proteins (BRD4, BRDT, BORIS and CTCF) indicated systematic highest binding when each energetic marks have been current and completely different selective binding when current alone at chromatin.
H3K27cr and H3K27ac lastly mark the constructing of some sperm super-enhancers. This built-in evaluation of omics information gives an unprecedented degree of understanding of gene expression regulation by H3K27cr compared to H3K27ac, and divulges each synergistic and particular actions of every histone modification.