Abstract
We present the design of three expression vectors that can be used for rapid cloning of any blunt-ended DNA segment. Only a single set of oligonucleotides is required to perform target DNA amplification and cloning into all three vectors simultaneously. The DNA thus cloned can express a protein with or without a hexahistidine tag depending on the vector used. The expression occurs from the T7 promoter when transformed into E. coli BL21(DE3).
Two of the three plasmids have been designed to provide the expressed protein with 6 N- or C-terminal 9xHis-tagged Recombinant amino acids in tandem. The third plasmid, however, does not add any tag to the expressed protein. Cloning is rapidly achieved with the requirement of phosphorylation of the PCR product without any restriction digestion. Furthermore, clones generated can be confirmed with a one-step PCR reaction performed from bacterial colonies (generally referred to as “colony PCR”).
We show the cloning, expression and purification of Green Fluorescent Protein (GFP) as a proof of concept. Furthermore, we also show the cloning and expression of four sigma factors from Mycobacterium tuberculosis, further demonstrating the utility of the engineered plasmids. We strongly believe that the vectors and strategy we have developed will facilitate the rapid cloning and expression of any gene in E. coli BL21(DE3) with or without a hexahistidine tag.
Materials and methods
- Bacterial strains, plasmids and growth conditions
Expression plasmids pET21b and pET15b were purchased from Novagen. EXPRESS chemically competent E. coli strains BL21(DE3) and XL1-blue were obtained from Lucigen and Stratagene respectively. E. coli bacterial strains were grown in Luria-Bertani medium (HiMedia), either as a liquid culture with constant shaking at 200 rpm or on a 1.5% agar plate at 37 °C. Cultures were always supplemented with 100 µg/ml ampicillin unless otherwise specified. All molecular biology methods and necessary precautionary measures were followed. Plasmid pSK01-NCHS was kindly provided by Soumya Kamilla, IISER Bhopal, India.
- Reagents
Restriction endonucleases, Antarctic phosphatase, and T4 polynucleotide kinase (PNK) were obtained from New England Biolabs (NEB) and used according to the manufacturer’s instructions. T4 DNA ligase was obtained from Fermentas and used as suggested. Plasmid DNA purification and DNA gel extraction kits were purchased from Qiagen and Sigma, respectively. The 2 log DNA ladder was obtained from NEB for DNA electrophoresis on agarose gels. The PiNK Plus protein ladder was purchased from GeneDireX and used according to instructions in acrylamide gel electrophoresis experiments. All other reagents were purchased from Sigma.
- GFP Expression and Zymography
Plasmids carrying the GFP gene were transformed into E. coli BL21(DE3) cells. Each clone was grown at 37°C at 200 rpm in 5 ml of LB medium. At an optical density at 600nm (OD600), ~0.8, 0.5 ml of culture was removed and 1 mM IPTG (final concentration) was added to the remaining media for protein expression. Cultures were allowed to grow further for 3 hours. The bacteria in all these cases were collected by centrifugation and 50 µl of 8 M urea and 10 µl of 5X SDS gel loading buffer were added to each.
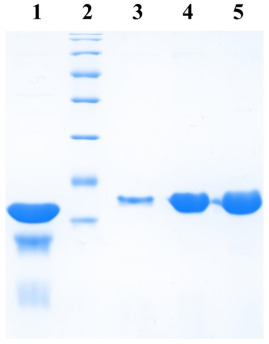
Cells were lysed by heating at 100°C for 5 min and 8 µl of each sample was subjected to 12% SDS-polyacrylamide gel electrophoresis (PAGE). After performing electrophoresis, the gel was incubated in 1% Triton X-100 for 2 hours and then photographed, to obtain a zymogram, with epi-illumination at 480 nm and SYBR Gold filter (485-655 nm) in a UVP. gel documentation system (UVP, LLC). Subsequently, the gel was fixed in a solution of 10% acetic acid, 1% trichloroacetic acid (TCA), and 40% methanol in water for 1 hour and then stained with Coomassie Brilliant Blue R-250 stain to visualize the protein bands.
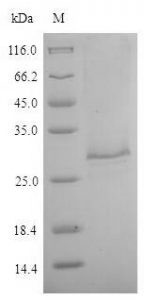
GFP purification
Clones expressing GFP along with the His6 tag were grown at 37°C at 200 rpm in 250 ml of LB medium. The protein was expressed by the addition of 1 mM IPTG when the culture reached OD600 ~0.8. The induction was continued for 3 hours. Cells were then collected by centrifugation at 4 °C and resuspended in the lysis buffer (50 mM sodium phosphate buffer, 500 mM NaCl, 5 mM β-mercaptoethanol, 10 mM imidazole, 5% glycerol) supplemented with 10 µg/ml lysozyme (chicken egg white lysozyme; Sigma) and incubated on ice for 30 min.
Cells were then lysed by sonication and the lysate cleared by centrifugation. The supernatant was incubated with 250 µl bed volume of Ni-NTA (Nickel-Nitrilotriacetic acid) agarose (Qiagen), pre-equilibrated with the lysis buffer, for 1 hour with constant mixing at 4°C. The matrix was collected and washed with wash buffer (50mM sodium phosphate buffer, 1M NaCl, 5mM β-mercaptoethanol, 20mM imidazole, 5% glycerol). Protein elution was then carried out in 10 column volumes of elution buffer (50 mM sodium phosphate buffer, 500 mM NaCl, 5 mM β-mercaptoethanol, 200 mM imidazole, 5% glycerol) and fractions elution collected were analyzed at a rate of 12%. SDS-PAGE.
Cloning of Sigma Factors from Mycobacterium tuberculosis and their Expression Analysis
Four extracytoplasmic sigma factors (SigB, D, F and G) from M. tuberculosis were cloned into the designed PMS-QS vectors. Genes were amplified by PCR using the genomic DNA of M. tuberculosis H37Rv as a template (kindly provided by AstraZeneca, Bangalore) and the primers. The PCR products were extracted from the agarose gel and ligated into the vectors. linear PMS-QS. . The resulting products were transformed into E. coli XL1Blue and positive clones were identified by colony PCR. All clones were further confirmed by sequencing. Plasmids so constructed were transformed into E. coli BL21 (DE3) for protein expression as described above.
Results
In the present manuscript, we report the design and construction of “rapid series” PMS vectors. These vectors contain a restriction enzyme site that blunts the vector for cloning of the DNA fragments generated by PCR. The vector carries a T7 promoter and a ribosome binding site upstream of the cloning site with the appropriate addition of codons for six histidine amino acids.